Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate Covid-19
Trial Design and Oversight
We conducted this three-group trial at 55 hospitals in Brazil. The trial was designed by the executive committee (see the Supplementary Appendix, available with the full text of this article at NEJM.org) and approved by the Brazilian National Commission for Research Ethics, the Brazilian Health Regulatory Agency (ANVISA), and ethics committees at the participating sites. The trial was funded by the hospitals and research institutes participating in Coalition Covid-19 Brazil (see the Supplementary Appendix). EMS Pharma provided additional funding and logistic support for the trial and also donated and supplied the trial drugs. EMS Pharma had no role in the conduct of the trial, the analysis, or the decision to submit the manuscript for publication. The trial was overseen by an independent international data and safety monitoring committee. The executive committee vouches for the completeness and accuracy of the data and for the fidelity of the trial to the protocol (available at NEJM.org).
Participants
The trial included consecutive patients who were 18 years of age or older and who had been hospitalized with suspected or confirmed Covid-19 with 14 or fewer days since symptom onset. Among the reasons for exclusion from the trial were the use of supplemental oxygen at a rate of more than 4 liters per minute as administered by a nasal cannula or at a level of at least 40% as administered by a Venturi mask; the use of supplemental oxygen administered by a high-flow nasal cannula or invasive or noninvasive ventilation; previous use of chloroquine, hydroxychloroquine, azithromycin, or any other macrolide for more than 24 hours before enrollment (and since the onset of symptoms); and a history of severe ventricular tachycardia or electrocardiographic findings with a corrected QT interval (QTc) of at least 480 msec. Complete information on the inclusion and exclusion criteria is provided in the Supplementary Appendix. All the patients provided written or electronic informed consent before randomization.
Randomization, Interventions, and Follow-up
Patients were randomly assigned in a 1:1:1 ratio to receive standard care (control group), standard care plus hydroxychloroquine at a dose of 400 mg twice daily for 7 days (hydroxychloroquine-alone group), or standard care plus hydroxychloroquine at a dose of 400 mg twice daily plus azithromycin at a dose of 500 mg once a day for 7 days. Randomization was performed in blocks of six and was stratified according to the use or nonuse of supplemental oxygen at the time of randomization. Randomization was performed centrally by means of an electronic case-report form system (RedCap) as described in the Supplementary Appendix.12
The current standard care for Covid-19 was at the discretion of the treating physicians. The use of glucocorticoids, other immunomodulators, antibiotic agents, and antiviral agents was allowed (see the Supplementary Appendix). The administration of hydroxychloroquine or chloroquine was not allowed in the control group, and the use of macrolides was not allowed in the control group or the hydroxychloroquine-alone group. Guidance was provided to the investigators about how to adjust or interrupt treatment according to side effects and laboratory abnormalities.
Data were collected daily, from randomization until day 15, in the electronic case-report form. For patients who were discharged before day 15, a structured telephone call to the patient or the patient’s family was conducted on or after day 15 by an interviewer who was unaware of the assigned trial group in order to assess vital status and return to routine activities.
Outcomes
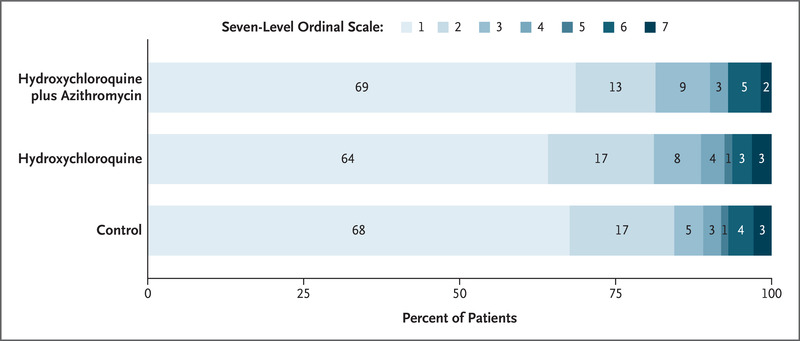
The primary outcome was clinical status at 15 days, evaluated with the use of a seven-level ordinal scale. Scores on the scale were defined as follows: a score of 1 indicated not hospitalized with no limitations on activities; 2, not hospitalized but with limitations on activities; 3, hospitalized and not receiving supplemental oxygen; 4, hospitalized and receiving supplemental oxygen; 5, hospitalized and receiving oxygen supplementation administered by a high-flow nasal cannula or noninvasive ventilation; 6, hospitalized and receiving mechanical ventilation; and 7, death.
Secondary outcomes included clinical status at 7 days, evaluated with the use of a six-level ordinal scale (see below and see the Supplementary Appendix); an indication for intubation within 15 days; the receipt of supplemental oxygen administered by a high-flow nasal cannula or noninvasive ventilation between randomization and 15 days; duration of hospital stay; in-hospital death; thromboembolic complications; acute kidney injury; and the number of days alive and free from respiratory support up to 15 days. A day alive and free from respiratory support was defined as any day in which the patient did not receive supplemental oxygen or invasive or noninvasive mechanical ventilation, from randomization to day 15. Patients who died during the 15-day window were assigned a value of 0 days alive and free from respiratory support in this assessment. Safety outcomes are listed in the Supplementary Appendix. All the trial outcomes were assessed by the site investigators, who were aware of the trial-group assignments (except as noted above for patients who had been discharged before day 15 and who were assessed for the primary outcome by means of a blinded telephone interview). No formal adjudication of trial outcomes was performed.
Sample-Size Calculation and Protocol Changes
We had originally planned for the trial to include 630 patients, using the intention-to-treat analysis population, with a six-level ordinal outcome as the primary outcome, as described in the Supplementary Appendix. However, before the first interim analysis was conducted, we changed the primary-outcome assessment to the seven-level ordinal scale and the main analysis population from the intention-to-treat population to a modified intention-to-treat population that included only patients with a diagnosis of Covid-19 that had been confirmed by reverse-transcriptase–polymerase-chain-reaction (RT-PCR) testing (using the test available at each site).
The change to the use of the seven-level ordinal scale was adopted because on April 10, 2020 (before the first enrolled patient had reached 15 days of follow-up), we established the capability to obtain 15-day information on limitations on activities with the use of blinded telephone interviews. We therefore added another level to the six-level ordinal outcome, dividing the first level (not hospitalized) into two levels (level 1, not hospitalized and with no limitations on activities; and level 2, not hospitalized but with limitations on activities). The change to the modified intention-to-treat population was adopted because, under the hypothesis that treatment would have beneficial effects on the primary outcome only for patients who had a confirmed diagnosis, the inclusion of unconfirmed cases would decrease the estimated effect size and power. As a related change, we added external adjudication of unconfirmed cases, which were classified as probable, possible, or probably not Covid-19 (see the Supplementary Appendix).
The sample size was revised with the use of the overall distribution of the seven-level ordinal outcome at day 15 observed among the first 120 patients, with the levels 1 through 7 having the following proportions of patients: 60%, 19%, 7%, 1%, 1%, 5%, and 7%, respectively. With 630 patients who had undergone randomization and 510 patients included in the modified intention-to-treat analysis, we calculated that the trial would have 80% power to detect an odds ratio of 0.5 between groups (two-by-two comparisons), at a significance level of 5% and with Bonferroni adjustment for multiple comparisons (α=5%, divided by 3 for each comparison).13
Statistical Analysis
The primary outcome was analyzed by mixed ordinal logistic regression with random intercept according to site, assuming proportional odds. We report all two-by-two comparisons. Binary outcomes were assessed with the use of a mixed logistic-regression model, except for in-hospital mortality, which was assessed with a Cox proportional-hazards model. Continuous outcomes were evaluated by means of generalized linear regression or mixed models for repeated variables, as appropriate. All models were adjusted for age and the use of supplemental oxygen at admission.
We also performed sensitivity analyses that included all the patients who had undergone randomization (intention-to-treat population) and sensitivity analyses for the primary outcome for the following groups: patients with definitive, probable, or possible Covid-19; and patients with definitive or probable Covid-19. Two additional populations were considered. An efficacy population included patients with a confirmed diagnosis who received at least one dose of the assigned trial drug. The safety population included patients according to the medications received, regardless of the assigned trial group or the result of Covid-19 testing.
We planned three interim analyses, to be conducted when 120 patients, 315 patients, and 504 patients had completed 15 days of follow-up. However, only the first interim analysis was conducted. Owing to faster-than-expected enrollment, primary-outcome data for the second and third interim analyses were available only after trial recruitment was finished. After discussion with the data and safety monitoring committee, the second and third interim analyses were cancelled. The data and safety monitoring committee used Haybittle–Peto14 stopping boundaries, with a P-value threshold of less than 0.001 to interrupt the trial for safety and a P-value threshold of less than 0.0001 to interrupt the trial for efficacy. We did not adjust the final values of the hypothesis test for sequential analyses.
Analyses were performed with the use of R software (R Core Team).15 P values for the primary outcome were adjusted with the use of Bonferroni correction. No P values are reported for secondary outcomes; the widths of the confidence intervals for the secondary outcomes have not been adjusted for multiple comparisons, so the intervals should not be used to infer definitive treatment effects. P values for the safety analyses were not adjusted given the importance of identifying potential signals of harm. Additional details about the statistical analyses are provided in the Supplementary Appendix.


